

BrainStorm (BCLI): Will the FDA Adcomm finally put it out of its misery?
BrainStorm (BCLI): Will the FDA Adcomm finally put it out of its misery?
And an update on Sage and a clearer picture on why they didn't get approved for MDD
BrainStorm Cell Therapeutics is one of those ludicrously bad stories in biotech that you’re amazed they are still around. They’ve never actually had a successful trial of NurOwn, their therapy for amyotrophic lateral sclerosis (ALS) that uses autologous mesenchymal stem cells to attempt to treat that horrible disease.
The results of their Phase 3 trial were not just bad but absolutely atrocious. Let’s dive in, shall we?:

As you can see, none of the primary endpoints were anywhere close to statistical significance, with one of the secondary endpoints demonstrating a p-value of 0.997. And then if you look at that secondary endpoint at the bottom, “event free probability for deaths due to disease progression”, you were more likely to die in the treatment arm due to disease progression, indicating that maybe the drug isn’t working.

And of course, just like crappy biotech’s have been doing for ages, they decided to rely on a subset of the data, in this case, those with an ALSFRS-R baseline score of greater than or equal to 35. Of course, even that data was negative in the original publication:
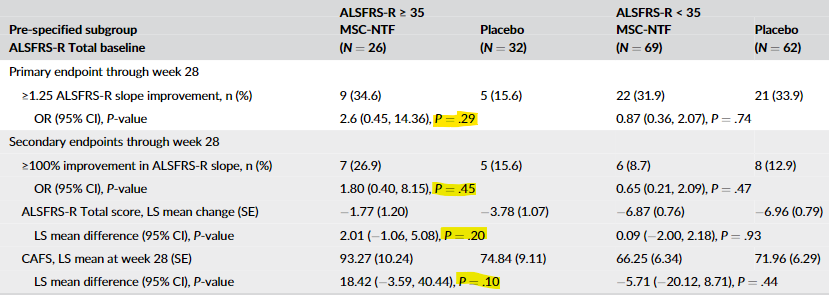
But then they “discovered” an error. They state in the erratum: “Subgroup analyses reported in the original publication for the key secondary endpoint, average change from baseline to endpoint in ALSFRS-R, used an incorrect model and incorporated interaction terms between the subgroup and treatment.” So now the second listed secondary endpoint in the above table looks like this:
As you can see, it now just squeaks through with a p-value of 0.05. But it appears the primary endpoint and the other two secondary endpoints are still quite negative even in this subgroup.
I’m sure some of you are saying “ALS is such a horrible disease, the FDA will surely bend over backwards to provide these patients with an option.” I don’t know about that, after the Phase 3 data was released, the FDA took the extremely unusual step of issuing a comment on the data (emphasis added by me):
With the recent completion of a randomized phase 3 controlled clinical trial comparing NurOwn to placebo, it has become clear that data do not support the proposed clinical benefit of this therapy. Data indicated that none of the primary or secondary endpoints were met in the group of patients who were randomized. For the main (primary) endpoint, 27.7% of people given the placebo were scored as responding compared to 32.6% of people given NurOwn. The 4.9% absolute difference in responders was not at all statistically significant, and the small difference between the two groups was most likely due to chance. In addition, there was a modest excess in deaths in those treated with NurOwn, the significance of which is unclear at this time. If BrainStorm plans further studies of NurOwn to determine if the product can provide clinical benefit to individuals with ALS, FDA will continue to provide advice to the company on their development program.
Now you’re probably saying, “the ALS community is so powerful that they will force the FDA to approve”. Here’s a comment from the ALS Association (which helped fund the Phase 3 trial) on NurOwn (again, emphasis added by me):
On June 6, 2023, BrainStorm announced that its advisory committee meeting (AdComm) for NurOwn will take place on September 27, 2023. It also announced that its PDUFA date, which is the date by which the FDA must decide whether to approve a treatment, is December 8, 2023
We only make recommendations on drug approvals after seeking independent peer review (as described here). Specifically, we ask sponsors to share data that will be submitted to the FDA and engage the recommendations of external experts who are not conflicted in any way with the program or the sponsor.
(Consistent with that policy, we consulted with independent experts and worked with Amylyx and Biogen to review FDA package data for AMX0035 (Relyvrio) and tofersen (Qalsody) before taking any stance on advocating at the AdComm and urging for their approval.)
After BrainStorm shared that its Phase 3 trial of NurOwn did not meet its primary or secondary endpoints, we have consistently requested access to the full data package so we could try to better understand its effect on people living with ALS. The amazing testimonials we have seen online do not align with the data that BrainStorm has shared with us or has been published in peer-reviewed publications.
We have an obligation to the community we serve to be vigilant and data-driven, and our approach has served the ALS community well in recent FDA reviews. Until we have the opportunity to conduct an independent review, we cannot take a position for or against approval of NurOwn. If Brainstorm wishes us to make any statements to the FDA, we encourage them to go through this independent review process.
It’s highly unusual for a company to piss off both the FDA and patient groups at the same time but somehow BrainStorm did it! It’s also a bit disconcerting that they aren’t willing to share the full data set which seems to imply they have something to hide.
Beyond efficacy, I suspect the FDA has other issues with NurOwn given what another mesenchymal stem cell company, Mesoblast, has had to deal with. Essentially, the FDA is unsure how you even quality control such a product. Here are comments from their August 13, 2020 adcomm (again, emphasis added):
However, as we heard in Dr. Temple's presentation this morning, MSCs are complex and have multimodal biological activities. Also, Dr. Temple's talk and the scientific literature describe considerable heterogeneity of MSCs due to differences in biological activity and donor-related effects. This raises the essence of the question we wish to discuss today, are the potency tests and other critical quality attributes for remestemcel-L sufficient to ensure the quality of the product from batch to batch?
And these issues STILL have not been resolved 3 years later (though the company states the assay has improved, the product has yet to be approved and the FDA is now requiring another clinical trial).
It’s therefore no surprise that after the company filed for approval in September of 2022, they received a refusal to file (RTF) letter from the FDA in November. Per the 10-K, the letter “included one item related to the trial not meeting the standard for substantial evidence of effectiveness and Chemistry, Manufacturing and Controls (“CMC”) related items.” And the only reason that the company is getting an adcomm at all is because they essentially demanded one. They requested the FDA file the application “over protest”, which they did. I don’t expect September 27th, the date of the adcomm, will be a good one for the company.
What’s next for the company if the adcomm is negative? It’s possible that they might finally be put out of their misery. As of June 30, 2023, they had $750,000 in cash and cash equivalents, with about $5.2 million in accounts payable and a quarterly cash burn from operations of $6.8 million. The only reason they aren’t bankrupt already is that they raised $7.5 million in gross proceeds on July 17 (probably a little under $7 million net after fees). Based on my calculations, they probably will only have 2-4 weeks of cash left when the FDA adcomm takes place. If it’s as negative as I expect, the company might be finished.
That said, advisory committee meetings can be quite emotional and they can turn on a dime, so I’ve seen some pretty strange things happen. But regardless of the outcome, I still wouldn’t expect FDA to approve on the PDUFA of December 8, 2023. Mesoblast, which I mentioned earlier, ended up having a positive FDA adcomm with a vote of 9-1 in favor, yet here we are 3 years later and there is no approval and they need to run another trial.
Of course you could feel bad for the company, but you probably shouldn’t. They’ve paid themselves VERY well, and mostly in cash rather than stock. In 2021, Chaim Lebovitz received $1.1 million in total compensation, with $750,000 (~68%) of that just in cash. Even worse is that the percent of his compensation coming from options that year was just under 10%. In comparison, in the S&P 500, typically 68% of CEO pay comes from stock and stock options and related instruments. Seems to me he didn’t really believe too much in his own company or he’d have more at-risk compensation.

Anyway, we’ll see how this plays out in a few weeks.
Sage’s Zurzuvae (zuranolone) is just too dangerous for a broad indication
As I mentioned previously, Sage Pharmaceuticals (SAGE) has a bit of a history of holding back bad news. Well, it looks like they are also not above using a “squirrel!” strategy of misdirection either.

On the exact same day that the FDA posted their review documents regarding Zurzuvae (zuranolone), Sage decided to announce their strategic reorganization of the company, which included a 40% workforce reduction and the departure of their Chief Scientific Officer and Chief Development Officer. This extends their cash runway until 2026. Now I’m not saying this wasn’t newsworthy but the timing is highly suspicious given the contents of the FDA review documents.
From what’s in the review documents it's pretty clear that Zurzuvae (zuranolone) has some significant safety issues. It caused confusion, seizures, psychosis and knocked patients unconscious. Here is a list of severe adverse events from the pivotal studies which occurred at the approved dosage of the medication:

Then there were some very serious cases in those who took supratherapeutic doses (which aren't necessarily that much higher than the approved 50mg dose). Here is someone who ended up being unconscious for 5 hours!:

Then here is someone who became unconscious twice!:

Another person had to go into a wheelchair:

Then someone who took the equivalent of the approved dose was aphasic (unable to comprehend or formulate language) for 45 minutes:

And here is someone with postpartum depression (PPD) who took 30mg and then got so confused they couldn’t take care of their baby!:

And pre-clinically the compound killed a number of dogs, likely due to physical dependence on the drug:

So an addictive drug that can knock you unconscious for hours? It’s a wonder the FDA even approved it for PPD given how dangerous it is (though that could have been political pressure) . One thing is clear, major depressive disorder is not going to be happening given this safety profile and meager efficacy. If they continue with that program, they might as well be burning cash in a bonfire.
Please like, share and subscribe!
Stocks mentioned: